
Regras de prioridade Cahn-Ingold-Prelog - Cahn–Ingold–Prelog priority rules

As regras de sequência Cahn-Ingold-Prelog ( CIP ) , nomeadas em homenagem aos químicos orgânicos Robert Sidney Cahn , Christopher Kelk Ingold e Vladimir Prelog - alternativamente chamadas de regras , sistema ou convenções de prioridade CIP - são um processo padrão usado na química orgânica para e nomear inequivocamente um estereoisômero de uma molécula. O objetivo do sistema CIP é atribuir um descritor R ou S a cada estereocentro e um descritor E ou Z a cada ligação dupla, de modo que a configuração de toda a molécula possa ser especificada exclusivamente, incluindo os descritores em seu nome sistemático. Uma molécula pode conter qualquer número de estereocentros e qualquer número de ligações duplas , e cada uma geralmente dá origem a dois isômeros possíveis. Uma molécula com um inteiro n que descreve o número de seus centros estereogênicos geralmente terá 2 n estereoisômeros e 2 n -1 diastereômeros, cada um tendo um par de enantiômeros associado. As regras de sequência CIP contribuem para a nomenclatura precisa de cada estereoisômero de cada molécula orgânica e organometálica com todos os átomos de ligância de menos de 4 (mas incluindo ligância de 6 também, este termo se referindo ao "número de átomos vizinhos" ligados a um Centro).
O artigo principal estabelecendo as regras de sequência CIP foi publicado em 1966, e foi seguido por outros refinamentos, antes de ser incorporado às regras da União Internacional de Química Pura e Aplicada (IUPAC), o órgão oficial que define a nomenclatura orgânica , em 1974. As regras foram revisadas, mais recentemente, em 2013, como parte do livro Nomenclature of Organic Chemistry da IUPAC . A apresentação das regras pela IUPAC constitui o padrão formal oficial para seu uso, e observa que "o método foi desenvolvido para cobrir todos os compostos com ligância até 4 ... e ... [estendido ao caso de] ligância 6 ... [bem como] para todas as configurações e conformações de tais compostos. " No entanto, embora a documentação da IUPAC apresente uma introdução completa, ela inclui o cuidado de que "é essencial estudar os artigos originais, especialmente o de 1966, antes de usar a regra de sequência para outros casos que não sejam simples."
Um artigo recente defende mudanças em algumas das regras (regras de sequência 1b e 2) para tratar de certas moléculas para as quais os descritores corretos não eram claros. No entanto, um problema diferente permanece: em casos raros, dois estereoisômeros diferentes da mesma molécula podem ter os mesmos descritores CIP, então o sistema CIP pode não ser capaz de nomear um estereoisômero de forma inequívoca, e outros sistemas podem ser preferíveis.
Passos para nomear
As etapas para nomear moléculas usando o sistema CIP são frequentemente apresentadas como:
- Identificação de estereocentros e duplas ligações ;
- Atribuição de prioridades aos grupos ligados a cada estereocentro ou átomo de ligação dupla; e
- Atribuição de descritores R / S e E / Z.
Atribuição de prioridades
Os descritores R / S e E / Z são atribuídos usando um sistema para classificar a prioridade dos grupos anexados a cada estereocentro. Esse procedimento, geralmente conhecido como regras de sequência , é o coração do sistema CIP. A visão geral nesta seção omite algumas regras que são necessárias apenas em casos raros.
- Compare o número atômico ( Z ) dos átomos diretamente ligados ao estereocentro; o grupo com o átomo de maior número atômico recebe maior prioridade.
- Se houver um empate, devemos considerar os átomos a uma distância 2 do estereocentro - já que uma lista é feita para cada grupo de átomos ligados àquele diretamente ligado ao estereocentro. Cada lista é organizada em ordem decrescente de número atômico. Em seguida, as listas são comparadas átomo a átomo; na primeira diferença, o grupo que contém o átomo de número atômico mais alto recebe prioridade mais alta.
- Se ainda houver um empate, cada átomo em cada uma das duas listas é substituído por uma sublista dos outros átomos ligados a ele (na distância 3 do estereocentro), as sublistas são organizadas em ordem decrescente de número atômico, e todo o a estrutura é novamente comparada átomo por átomo. Este processo é repetido recursivamente, cada vez com os átomos uma ligação mais longe do estereocentro, até que a ligação seja quebrada.
Isótopos
Se dois grupos diferem apenas em isótopos , a massa atômica maior é usada para definir a prioridade.
Ligações duplas e triplas

Se um átomo A é duplamente ligado a um átomo B, A é tratado como tendo uma ligação simples a dois átomos: B e um "átomo fantasma" que é uma duplicata de B (tem o mesmo número atômico), mas não está ligado a nada exceto A. Quando B é substituído por uma lista de átomos anexados, o próprio A, mas não seu "fantasma", é excluído de acordo com o princípio geral de não retroceder ao longo de uma ligação que acabou de ser seguida. Uma ligação tripla é tratada da mesma maneira, exceto que A e B estão conectados a dois átomos fantasmas um do outro.
Isômeros geométricos
Se dois substituintes em um átomo são isômeros geométricos um do outro, o isômero Z tem maior prioridade do que o isômero E.
Moléculas cíclicas
Para lidar com uma molécula contendo um ou mais ciclos , deve-se primeiro expandi-la em uma árvore (chamada dígrafo hierárquico ), percorrendo ligações em todos os caminhos possíveis, começando no estereocentro. Quando a travessia encontra um átomo através do qual o caminho atual já passou, um átomo fantasma é gerado para manter a árvore finita. Um único átomo da molécula original pode aparecer em muitos lugares (alguns como fantasmas, outros não) na árvore.
Atribuição de descritores
Estereocentros: R / S

Após os substituintes de um estereocentro terem sido atribuídos às suas prioridades, a molécula é orientada no espaço de forma que o grupo com a prioridade mais baixa seja apontado para longe do observador. Se os substituintes forem numerados de 1 (prioridade mais alta) a 4 (prioridade mais baixa), então o sentido de rotação de uma curva que passa por 1, 2 e 3 distingue os estereoisômeros . Um centro com sentido de rotação no sentido horário é um centro R ( reto ) e um centro com sentido de rotação no sentido anti-horário é um centro S ( sinistro ). Os nomes são derivados do latim para 'direita' e 'esquerda', respectivamente.
-1,2,3-trichlorocyclopentane.svg)
Um método prático para determinar se um enantiômero é R ou S é usando a regra da mão direita : envolve a molécula com os dedos na direção 1 → 2 → 3 . Se o polegar apontar na direção do quarto substituinte, o enantiômero é R ; caso contrário, é S .
Em casos raros, é possível que dois substituintes em um átomo difiram apenas em sua configuração absoluta ( R ou S ). Se as prioridades relativas destes substituintes precisam ser estabelecidos, R tem prioridade sobre S . Quando isso acontece, o descritor do estereocentro é uma letra minúscula ( r ou s ) em vez da letra maiúscula normalmente usada.
Ligações duplas: E / Z
Para alcenos e moléculas de ligação dupla semelhantes, o mesmo processo de priorização é seguido para os substituintes. Neste caso, é a colocação dos dois substituintes de maior prioridade em relação à ligação dupla que importa. Se ambos os substituintes de alta prioridade estiverem no mesmo lado da ligação dupla, isto é, na configuração cis , então o estereoisômero é designado como Z ( zusammen ). Se, pelo contrário, estiverem em uma configuração trans , o estereoisômero é atribuído a um E ( entgegen ). Neste caso, as letras de identificação são derivadas do alemão para 'junto' e 'oposto', respectivamente.
Exemplos
A seguir estão exemplos de aplicação da nomenclatura.
Atribuições R / S para vários compostos 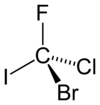
A molécula hipotética bromoclorofluoroiodometano mostrada em sua configuração ( R ) seria um composto quiral muito simples. As prioridades são atribuídas com base no número atômico ( Z ): iodo ( Z = 53)> bromo ( Z = 35)> cloro ( Z = 17)> flúor ( Z = 9). Permitindo que o flúor (prioridade mais baixa) aponte para longe do visualizador, a rotação é no sentido horário, portanto, a atribuição R. 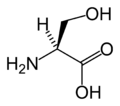
Na atribuição de L- serina, a prioridade mais alta é dada ao átomo de nitrogênio ( Z = 7) no grupo amino (NH 2 ). Tanto o grupo hidroximetil (CH 2 OH) quanto o grupo de ácido carboxílico (COOH) têm átomos de carbono ( Z = 6), mas a prioridade é dada ao último porque o átomo de carbono no grupo COOH está conectado a um segundo oxigênio ( Z = 8 ), enquanto no grupo CH 2 OH o carbono está ligado a um átomo de hidrogênio ( Z = 1). Mais baixa prioridade é dada para o átomo de hidrogénio e como esta pontos átomo de distância a partir do telespectador o decréscimo em sentido anti-horário prioridade sobre os restantes três substituintes completa a atribuição como S . 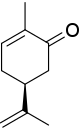
O estereocentro em ( S ) -carvona está conectado a um átomo de hidrogênio (não mostrado, prioridade 4) e três átomos de carbono. O grupo isopropenil tem prioridade 1 (átomos de carbono apenas) e para os dois átomos de carbono restantes a prioridade é decidida com os átomos de carbono duas ligações removidas do estereocentro, uma parte do grupo ceto (O, O, C, prioridade 2) e uma parte de um alceno (C, C, H, prioridade 3). Os resultados resultantes rotação anti-horária em S .


-Carvone.svg)
Descrevendo vários centros
Se um composto tem mais do que um estereocentro quiral, cada centro é denotada por um ou outro de R ou S . Por exemplo, a efedrina existe nos estereoisômeros (1 R , 2 S ) e (1 S , 2 R ), que são formas espelhadas distintas entre si, tornando-os enantiômeros . Este composto também existe como os dois enantiômeros escritos (1 R , 2 R ) e (1 S , 2 S ), que são chamados de pseudoefedrina em vez de efedrina. Todos os quatro destes isômeros são nomeados 2-metilamino-1-fenil-1-propanol na nomenclatura sistemática. No entanto, a efedrina e a pseudoefedrina são diastereômeros ou estereoisômeros que não são enantiômeros porque não estão relacionados como cópias de imagem em espelho. A pseudoefedrina e a efedrina recebem nomes diferentes porque, como diastereômeros, têm propriedades químicas diferentes, mesmo para misturas racêmicas de cada uma.
Mais geralmente, para qualquer par de enantiômeros, todos os descritores são opostos: ( R , R ) e ( S , S ) são enantiômeros, como são ( R , S ) e ( S , R ). Os diastereômeros têm pelo menos um descritor em comum; por exemplo ( R , S ) e ( R , R ) são diasteriômeros, assim como ( S , R ) e ( S , S ). Isso é válido também para compostos com mais de dois estereocentros: se dois estereoisômeros têm pelo menos um descritor em comum, eles são diastereômeros. Se todos os descritores forem opostos, eles são enantiômeros.
Quando a atribuição numérica dos estereocentros não é única por causa da simetria do espelho de toda a molécula, o resultado é um composto meso , como o ácido mesotartárico , no qual ( R , S ) é o mesmo que a forma ( S , R ). Em compostos meso, os estereocentros R e S ocorrem em pares posicionados simetricamente.
Configuração relativa
A configuração relativa de dois estereoisômeros pode ser denotada pelos descritores R e S com um asterisco (*). ( R *, R *) significa dois centros com configurações idênticas, ( R , R ) ou ( S , S ); ( R *, S *) significa dois centros com configurações opostas, ( R , S ) ou ( S , R ). Para começar, o centro estereogênico de numeração mais baixa (de acordo com a numeração sistemática da IUPAC) recebe o descritor R *.
Para designar dois anômeros, os estereodescritores relativos alfa (α) e beta (β) são usados. No anômero α, o átomo de carbono anomérico e o átomo de referência têm configurações opostas ( R , S ) ou ( S , R ), enquanto no anômero β são iguais ( R , R ) ou ( S , S ).
Rostos

A estereoquímica também desempenha um papel na atribuição de faces às moléculas trigonais, como as cetonas . Um nucleófilo em uma adição nucleofílica pode se aproximar do grupo carbonila de dois lados ou faces opostas. Quando um nucleófilo aquiral ataca a acetona , ambas as faces são idênticas e há apenas um produto de reação. Quando o nucleófilo ataca a butanona , as faces não são idênticas ( enantiotópicas ) e o resultado é um produto racêmico . Quando o nucleófilo é uma molécula quiral , diastereoisômeros são formados. Quando uma face de uma molécula é protegida por substituintes ou restrições geométricas em comparação com a outra face, as faces são chamadas de diastereotópicas . As mesmas regras que determinam a estereoquímica de um estereocentro ( R ou S ) também se aplicam ao atribuir a face de um grupo molecular. As faces são então chamadas de Re- face e Si -face . No exemplo exibido à direita, o composto acetofenona é visualizado no Re -face. A adição de hidreto como em um processo de redução deste lado formará o ( S ) -enantiômero e o ataque da face Si oposta dará o ( R ) -enantiômero. No entanto, deve-se observar que adicionar um grupo químico ao centro proquiral do Re- face nem sempre levará a um estereocentro ( S ), pois a prioridade do grupo químico deve ser levada em consideração. Ou seja, a estereoquímica absoluta do produto é determinada por si mesmo e não considerando de qual face ele foi atacado. No exemplo mencionado acima, se o cloreto ( Z = 17) fosse adicionado ao centro pró-quiral da Re- face, isso resultaria em um ( R ) -enantiômero.