
Borylation - Borylation
As reações de borilação C – H catalisadas por metal são reações orgânicas catalisadas por metal de transição que produzem um composto organoboro por meio da funcionalização de ligações C – H alifáticas e aromáticas e, portanto, são reações úteis para a ativação de ligações carbono – hidrogênio . As reações de boryilação C – H catalisadas por metal utilizam metais de transição para converter diretamente uma ligação C – H em uma ligação C – B. Esta rota pode ser vantajosa em comparação com as reações de borylation tradicionais, fazendo uso de material de partida de hidrocarboneto barato e abundante, limitando compostos orgânicos pré-funcionalizados, reduzindo subprodutos tóxicos e agilizando a síntese de moléculas biologicamente importantes. Ácidos borônicos e ésteres borônicos são grupos comuns de borila incorporados em moléculas orgânicas por meio de reações de borilação. Os ácidos borônicos são compostos orgânicos trivalentes contendo boro que possuem um substituinte alquil e dois grupos hidroxila. Da mesma forma, os ésteres borônicos possuem um substituinte alquil e dois grupos éster. Os ácidos e ésteres borônicos são classificados dependendo do tipo de grupo de carbono (R) diretamente ligado ao boro, por exemplo, alquil-, alquenil-, alquinil- e ésteres aril-borônicos. O tipo mais comum de materiais de partida que incorporam ésteres borônicos em compostos orgânicos para reações de boryilação catalisadas por metais de transição têm a fórmula geral (RO) 2 B-B (OR) 2 . Por exemplo, bis (pinacolato) diboro (B 2 Pino 2 ) e bis (catecolato) diborano (B 2 Cat 2 ) são fontes comuns de boro desta fórmula geral.

O átomo de boro de um éster ou ácido borônico é hibridizado com sp 2 possuindo um orbital p vago, permitindo que esses grupos atuem como ácidos de Lewis . A ligação C – B dos ácidos borônicos e ésteres é ligeiramente mais longa do que as ligações simples C – C com um intervalo de 1,55-1,59 Å. A ligação C – B alongada em relação à ligação C – C resulta em uma energia de ligação que também é ligeiramente menor do que a das ligações C – C (323 kJ / mol para C – B vs 358 kJ / mol para C – C). A ligação carbono-hidrogênio tem um comprimento de ligação de cerca de 1,09 Å e uma energia de ligação de cerca de 413 kJ / mol. A ligação C – B é, portanto, um intermediário útil como uma ligação que substitui uma ligação C – H tipicamente não reativa.
Os compostos de organoboro são compostos orgânicos que contêm uma ligação carbono-boro. Os compostos de organoboro têm amplas aplicações para síntese química porque a ligação C – B pode ser facilmente convertida em uma ligação C – X (X = Br, Cl), C – O, C – N ou C – C. Devido à versatilidade da ligação C – B, vários processos foram desenvolvidos para incorporá-los em compostos orgânicos. Os compostos de organoboro são tradicionalmente sintetizados a partir de reagentes de Grignard por meio de reações de hidroboração ou diboração. A borylation oferece uma alternativa.
Reações de borilação C – H catalisadas por metal
Alifático C-H borylation
Conforme descrito pela primeira vez por Hartwig, os alcanos podem ser borilados seletivamente com alta seletividade para a ligação C – H primária usando Cp * Rh (η 4 -C 6 Me 6 ) como o catalisador. Notavelmente, a seletividade para a ligação C – H primária é exclusiva, mesmo na presença de heteroátomos na cadeia de carbono-hidrogênio. A borilação catalisada por ródio de ligações metil C – H ocorre seletivamente, sem dependência da posição do heteroátomo. A borilação ocorre seletivamente pelo menos estereoquimicamente impedida e menos rica em elétrons na ligação C – H primária em uma gama de acetais , éteres , aminas e fluoretos de alquil. Além disso, nenhuma reação ocorre na ausência de ligações C – H primárias, por exemplo, quando o ciclohexano é o substrato.

A funcionalização seletiva de uma ligação alcano primário é devida à formação de um complexo alquil-metal primário cinética e termodinamicamente favorável sobre a formação de um complexo alquil-metal secundário.

A maior estabilidade de complexos de alquil primários versus secundários pode ser atribuída a vários fatores. Em primeiro lugar, o complexo de alquil primário é favorecido estericamente em relação ao complexo de alquil secundário. Em segundo lugar, cargas negativas parciais estão frequentemente presentes no carbono α de um complexo metal-alquil e um ligante de alquil primário suporta uma carga negativa parcial melhor do que um ligante de alquil secundário. A origem da seletividade para a boryilação alifática de C-H usando catalisadores de ródio foi sondada usando um tipo de estudo mecanístico chamado troca de hidrogênio-deutério . H / D trocado mostrou que a regiosseletividade do processo geral mostrado abaixo resulta da clivagem seletiva das ligações C-H primárias sobre as secundárias e da funcionalização seletiva do intermediário metal-alquil primário sobre o intermediário metal-alquil secundário.

A utilidade sintética da borilação C – H alifática foi aplicada à modificação de polímeros por meio de borilação seguida de oxidação para formar polímeros funcionalizados com hidroxila.

Borilação C – H aromática
Borilação C – H de arenos dirigida estérica
O primeiro exemplo de uma borilação C – H catalítica de um hidrocarboneto não ativado (benzeno) foi relatado por Smith e Iverson usando Ir (Cp *) (H) (Bpin) como o catalisador. A eficiência deste sistema, entretanto, foi baixa, proporcionando apenas 3 turnovers após 120 ha 150 ° C. Numerosos desenvolvimentos subsequentes por Hartwig e colegas de trabalho levaram a condições práticas e eficientes para a borilação de areno. A borilação aromática C – H foi desenvolvida por John F. Hartwig e Ishiyama usando o reagente diboro Bis (pinacolato) diboro catalisado por 4,4'-di-terc-butilbipiridina (dtbpy) e [Ir (COD) (OMe)] 2 . Com este sistema de catalisador, a borilação de ligações C – H aromáticas ocorre com regiosseletividade que é controlada por efeitos estéricos do areno inicial. A seletividade para funcionalização de ligações C – H aromáticas é governada pela regra geral de que a reação não ocorre orto para um substituinte quando uma ligação C – H sem um substituinte orto está disponível. Quando apenas um grupo funcional está presente, a boryilação ocorre nas posições meta e para em razões estatísticas de 2: 1 (meta: para). O isômero orto não é detectado devido aos efeitos estéricos do substituinte.

A adição de Bpin ocorre em apenas uma posição para arenos substituídos simetricamente 1,2 e 1,4. Os arenos 1,3-substituídos simétricos ou assimétricos também são borilados seletivamente porque apenas uma ligação C – H é estericamente acessível.

Isso contrasta com a substituição eletrofílica aromática, em que a regiosseletividade é governada por efeitos eletrônicos.

A importância sintética da borilação C – H aromática é mostrada abaixo, onde um composto aromático 1,3-dissubstituído pode ser convertido diretamente em um composto 1,3,5-organoborano e subsequentemente funcionalizado.

Aromático C-H funcionalização foi incorporado com sucesso na síntese total de Complanadine A, um Licopódio alcalóide que aumenta o ARNm de express para o factor de crescimento do nervo (NGF) e a produção de NGF em humanos células gliais . Produtos naturais que promovem o crescimento de novas redes neurais são de interesse no tratamento de doenças como a doença de Alzheimer . A complanadina A foi sintetizada com sucesso usando uma combinação de borylation direta C-H aromática desenvolvida por Hartwig e Ishyiama, seguido por acoplamento cruzado Suzuki-Miyaura e , em seguida, clivagem do grupo protetor Boc .

Borilação C – H de heteroarenos
Os heteroarenos também podem sofrer borylation sob condições catalisadas por irídio, no entanto, a seletividade do local, neste caso, é controlada por efeitos eletrônicos , onde furanos , pirróis e tiofenos sofrem reação na ligação C – H alfa para o heteroátomo. Nesse caso, sugere-se que a seletividade ocorra através da ligação C – H alfa para o heteroátomo porque é a ligação C – H mais ácida e, portanto, a mais reativa.

Directed orto C-H borylation
Usando o mesmo sistema de catalisador, grupos de direcionamento podem ser empregados para atingir regiosseletividade sem substituintes como mediadores estéricos. Por exemplo, Boebel e Hartwig relataram um método para conduzir orto- borilação, onde um grupo de direção de dimetil-hidrossilil no areno sofre boryilação catalisada por irídio na ligação C – H orto ao grupo de direção de silano . A seletividade para a posição orto no caso de usar grupos diretores de hidrossilila foi atribuída à adição reversível da ligação Si-H ao centro de metal, levando à clivagem preferencial da ligação C – H orto ao substituinte hidrossilila. Várias outras estratégias para alcançar a orto- borilação de arenos foram desenvolvidas usando vários grupos de direção.

Detalhe mecanístico para a boração C – H de arenos
Um complexo de trisboryl irídio foi proposto para facilitar o mecanismo para cada uma dessas reações que resultam na borilação C – H de arenos e heteroarenos. Estudos cinéticos e estudos de marcação isotópica revelaram que um complexo tri- boril Ir (III) reage com o areno no processo catalítico. Uma versão do ciclo catalítico é mostrada abaixo para a orto borilação de compostos de hidrossilano. Os dados cinéticos mostram que um complexo de trisboryl observado, coordenado ao cicloocteno, se dissocia rápida e reversivelmente do cicloocteno para formar um complexo de trisboryl de 16 elétrons. No caso do uso de benzildimetilsilano como um grupo direcionador, é proposto que o benzildimetilsilano reage com o catalisador de trisboryil irídio por meio da adição reversível da ligação Si-H ao centro do metal, seguida por ativação seletiva da ligação orto -C – H via adição oxidativa e redutiva eliminação .

Borylation meta-seletiva : a borylation C – H meta-seletiva é uma transformação sintética importante, que foi descoberta em 2002 por Smith III da Michigan State University, EUA. No entanto, esta metaborilação foi completamente estericamente dirigida e limitada a apenas benzenos 1,3-dissubstituídos. Cerca de 12 anos depois, o Dr. Chattopadhyay e sua equipe do Centro de Pesquisa Biomédica, UP, Índia, descobriram uma tecnologia elegante para a ativação e borilação da ligação C – H meta-seletiva. A equipe mostrou que usando o mesmo substrato, pode-se trocar a seletividade posicional apenas trocando o ligante. A origem da meta-seletividade foi definida por dois parâmetros, tais como: 1) interação eletrostática, 2) uma interação BN secundária.
Ao mesmo tempo, uma equipe do Japão, Dr. Kanai, relatou um conceito incrível para a borylation meta-seletiva com base na interação secundária. Este método cobre vários compostos de carbonila borylation.
Reações de redução com compostos de organoboro
Redução Corey-Bakshi-Shibata (redução CBS)
Em 1981, Hirao e colaboradores descobriram que a redução assimétrica de cetonas aromáticas próquirais com aminoálcoois quirais e borano proporcionou os álcoois secundários correspondentes com 60% ee . Eles descobriram que os aminoácidos quirais álcoois iria reagir com borano para formar complexos aloxyl-amina-borano. Propõe-se que os complexos contenham um sistema de anéis de cinco membros relativamente rígido, o que os torna estáveis termicamente e hidroliticamente e solúveis em uma ampla variedade de solventes próticos e apróticos.
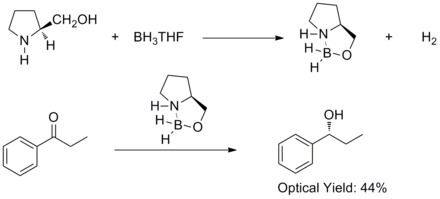
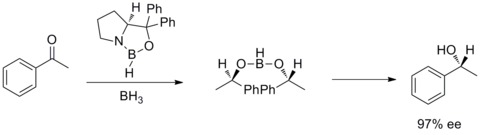
Em 1987, Elias James Corey e colaboradores descobriram a formação de oxazaborolidinas a partir de borano e aminoálcoois quirais . E as oxazaborolidinas catalisam a redução rápida e altamente enantiosseletiva de cetonas pró-quirais na presença de BH3THF. Essa redução enantiosseletiva de cetonas aquirais com oxazaborolidina catalítica é chamada de redução de Corey-Bakshi-Shibata ou redução de CBS.
Redução de Midland Alpine-borano (redução de Midland)
Em 1977, MM Midland e colegas de trabalho relataram uma observação surpreendente de que B-3-alfa-Pinanil-9-borabiciclo [3,3,1] nonano, prontamente preparado por hidroboração de (+) - alfa-pineno com 9-borobiciclo [3,3,1] nonano , reduz rapidamente o benzaldeído-alfa-d a álcool (S) - (+) - benzil-alfa-d com uma indução assimétrica essencialmente quantitativa.
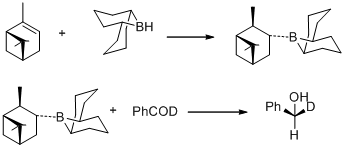
No mesmo ano, MM Midland descobriu o B-3-alfa-pinanil-9-BBN como o agente redutor, que poderia estar facilmente disponível ao reagir (+) - alfa-pineno com 9-BBN. O novo agente redutor foi posteriormente comercializado pela Aldrich Co. sob o nome de Alpine Borane e a redução assimétrica de grupos carbonil com qualquer enantiômero de Alpine-Borane é conhecida como redução Midland Alpine-Borane.
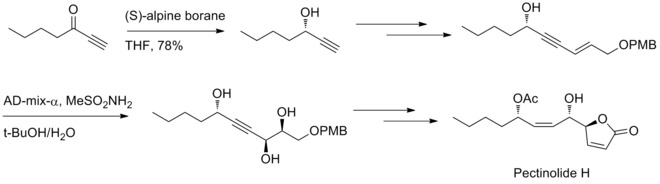
Em 2012, URY Venkateswarlu e colaboradores relataram um método estereosseletivo para sintetizar a redução do pectinolídeo H. Midland e a reação de diidroxilação de Sharpless estão envolvidos na geração dos três centros quirais em C – 4 ', C – 5 e C – 1'.
Reações de acoplamento com compostos de organoboro
Reação de ácido borônico Petasis-Mannich
Em 1993, NA Petasis e I. Akrltopoulou relataram uma síntese eficiente de aminas alílicas com uma reação de Mannich modificada . Nessa reação de Mannich modificada, eles descobriram que os ácidos vinilborônicos podem participar como nucleófilos para dar alilaminas geometricamente puras. Esta reação de Mannich modificada era conhecida como reação de Petasis ácido borônico-Mannich.
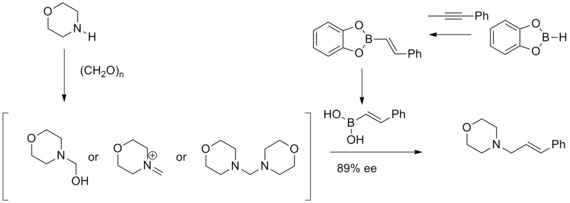
Alilação assimétrica de Roush
Em 1978, RW Hoffmann e T. Herold relataram a síntese enantiosseletiva de álcoois homoalílicos secundários via ésteres alilborônicos quirais não racêmicos . Os álcoois homoalílicos foram formados com excelente rendimento e moderada enantiosseletividade.
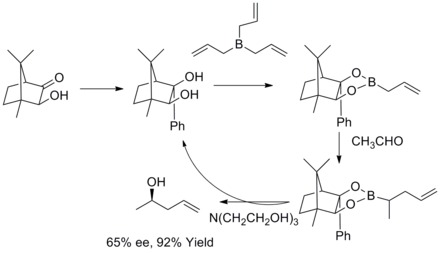
Em 1985, WR Roush e colaboradores descobriram que os boronatos alílicos modificados com tartarato oferecem uma abordagem simples e altamente atrativa para o controle da seletividade facial em reações com aldeídos quirais e aquirais. Nos anos seguintes, WR Roush e colaboradores estenderam essa estratégia para a síntese de but-2-eno-1,4-dióis e anti-dióis . Esse tipo de reação é conhecido como alilação assimétrica de Rouch.
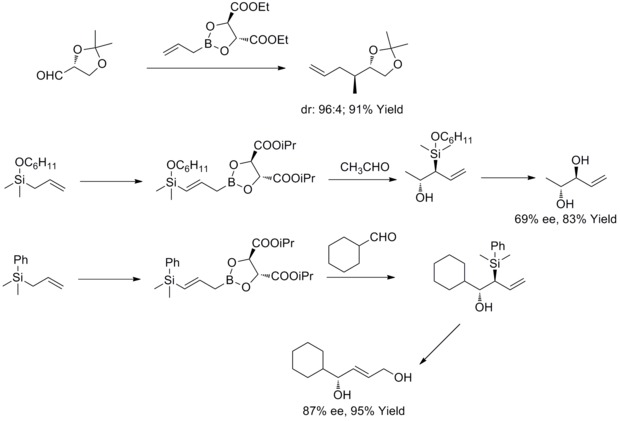
Em 2011, RA Fernandes e P. Kattanguru concluíram uma síntese total aprimorada de diastereômeros (8S, 11R, 12R) - e (8R, 11R, 12R) -topsentolídeo B2 em oito etapas. No artigo, a reação de alilação de Roush diastereosseletiva foi usada como uma reação chave na síntese total para introduzir dois intermediários quirais. E então os autores sintetizaram os dois diastereômeros por meio desses dois intermediários quirais.

Acoplamento cruzado Suzuki-Miyaura
Em 1979, N. Miyaura e A. Suzuki relataram a síntese de (E) -alcenos arilados em alto rendimento a partir de haletos de arila com alquil-1-enilboranos e catalisados por tetraquis ( trifenilfosfina ) paládio e bases. Então A. Suzuki e colegas de trabalho estendem este tipo de reação a outros compostos de organoboro e outros alquenil, aril , halogenetos de alquil e triflato . Os compostos de organoboro da reação de acoplamento cruzado catalisado por paládio e esses haletos orgânicos para formar ligações carbono-carbono são conhecidos como acoplamento cruzado de Suzuki-Miyaura .
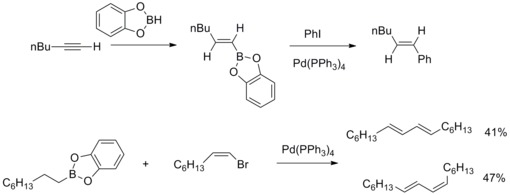
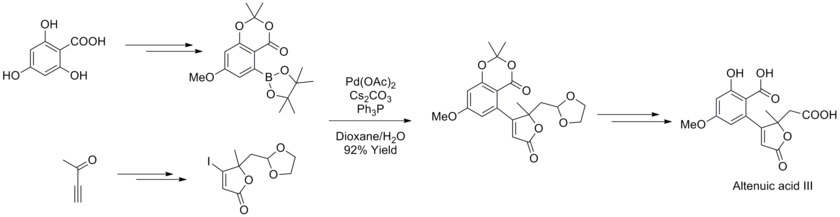
Em 2013, Joachim Podlech e colaboradores determinaram a estrutura da micotoxina ácido altenuico III de Alternaria por análise espectroscópica de RMN e completaram sua síntese total. Na estratégia sintética, a reação de acoplamento cruzado Suzuki-Miyaura foi usada com um boronato e butenolídeos altamente funcionalizados para sintetizar um precursor do produto natural em alto rendimento.
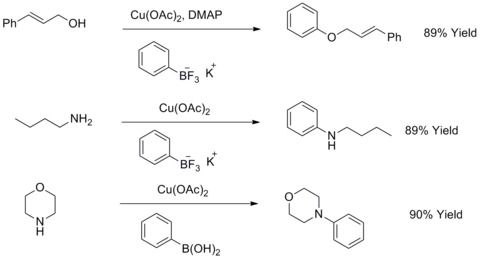
Síntese de éter biaril Ullmann modificado e biarilamina
Em 1904, Fritz Ullmann descobriu que o pó de cobre poderia melhorar significativamente a reação de haletos de arila com fenóis para dar éteres biarílicos. Esta reação é conhecida como condensação de Ullmann . Em 1906, I. Goldberg estendeu essa reação para sintetizar uma arilamina pela reação de haletos de arila com uma amida na presença de carbonato de potássio e CuI. Esta reação é conhecida como condensação de Ullmann modificada por Goldberg. Em 2003, RA Batey e TD Quach modificaram este tipo de reações usando sais organotrifluoroboratos de potássio para reagir com álcoois alifáticos, aminas alifáticas ou anilinas para sintetizar éteres arílicos ou arilaminas.
Veja também
- Química Organoboron
- Reações de organoboratos e boranos
- Redução Corey-Itsuno
- Redução de borano em Midland Alpine
- Reação de Petasis
- Reação de Suzuki