
Cicloadição 1,3-dipolar - 1,3-Dipolar cycloaddition
| Cicloadição 1,3-dipolar de Huisgen | |
|---|---|
| Nomeado após | Rolf Huisgen |
| Tipo de reação | Reação de formação de anel |
| Identificadores | |
| Portal de Química Orgânica | huisgen-1,3-dipolar-cicloadição |
| ID de ontologia RSC | RXNO: 0000018 |
A cicloadição 1,3-dipolar é uma reação química entre um 1,3-dipolo e um dipolarófilo para formar um anel de cinco membros. As primeiras cicloadições 1,3-dipolares foram descritas no final do século 19 até o início do século 20, após a descoberta dos 1,3-dipolos. A investigação mecanística e a aplicação sintética foram estabelecidas na década de 1960, principalmente por meio do trabalho de Rolf Huisgen . Portanto, a reação é algumas vezes referida como cicloadição de Huisgen (este termo é frequentemente usado para descrever especificamente a cicloadição 1,3-dipolar entre uma azida orgânica e um alcino para gerar 1,2,3-triazol ). A cicloadição 1,3-dipolar é uma rota importante para a síntese regio e estereosseletiva de heterociclos de cinco membros e seus derivados acíclicos de anel aberto. O dipolarófilo é tipicamente um alceno ou alcino, mas pode ser outros sistemas pi. Quando o dipolarófilo é um alcino, geralmente são produzidos anéis aromáticos.

Visão geral mecanicista
Originalmente, dois mecanismos propostos descrevem a cicloadição 1,3-dipolar: primeiro, o mecanismo de cicloadição pericíclico combinado , proposto por Rolf Huisgen; e segundo, o mecanismo passo a passo envolvendo um intermediário diradical , proposto por Firestone. Depois de muito debate, a primeira proposta agora é geralmente aceita - o 1,3-dipolo reage com o dipolarófilo de uma forma combinada , frequentemente assíncrona e simetria - permitida π 4 s + π 2 s por meio de uma transição aromática de Huckel de seis elétrons estado . No entanto, existem alguns exemplos de um mecanismo passo a passo para as reações de cicloadição 1,3-dipolar livres de catalisador de iletos de tiocarbonil e óxidos de nitrila

Mecanismo pericíclico
Huisgen investigou uma série de cicloadições entre os compostos 1,3-dipolares diazo e vários alcenos dipolarofílicos . As observações a seguir apóiam o mecanismo pericíclico combinado e refutam o dirradical gradativo ou a via polar gradativa.
- Efeitos do substituinte : Diferentes substituintes no dipolo não exibem um grande efeito na taxa de cicloadição, sugerindo que a reação não envolve um intermediário separado por carga.
- Efeitos do solvente : A polaridade do solvente tem pouco efeito na taxa de cicloadição, em linha com o mecanismo pericíclico, onde a polaridade não muda muito ao passar dos reagentes para o estado de transição.
- Estereoquímica : as cicloadições 1,3-dipolares são sempre estereoespecíficas com respeito ao dipolarófilo (isto é, cis- alcenos dando sin- produtos), apoiando o mecanismo pericíclico combinado no qual duas ligações sigma são formadas simultaneamente.
- Parâmetros termodinâmicos : as cicloadições 1,3-dipolares têm uma entropia negativa invulgarmente grande de ativação semelhante à da reação de Diels-Alder , sugerindo que o estado de transição é altamente ordenado, o que é uma assinatura de reações pericíclicas combinadas.
1,3-dipolo

Um 1,3-dipolo é uma molécula orgânica que pode ser representada como um tipo alil ou um octeto / sexteto zwitteriônico do tipo propargil / alenil . Ambos os tipos de 1,3-dipolo compartilham quatro elétrons no sistema π ao longo de três átomos. O tipo alil é curvado enquanto o tipo propargil / alenil é linear em geometria . 1,3-dipolos contendo elementos de linha superior, como enxofre ou fósforo, também são conhecidos, mas são utilizados com menos frequência.
Estruturas de ressonância podem ser desenhadas para deslocar cargas negativas e positivas em qualquer terminal de um 1,3-dipolo (veja o esquema abaixo). Um método mais preciso para descrever a distribuição eletrônica em um 1,3-dipolo é atribuir o principal contribuidor de ressonância com base em dados experimentais ou teóricos, como medições de momento dipolo ou cálculos. Por exemplo, o diazometano carrega o maior caractere negativo no átomo de nitrogênio terminal , enquanto o ácido hidrazóico carrega o maior caractere negativo no átomo de nitrogênio interno .

Consequentemente, essa ambivalência significa que as extremidades de um 1,3-dipolo podem ser tratadas como nucleofílicas e eletrofílicas ao mesmo tempo. A extensão da nucleofilicidade e eletrofilicidade em cada extremidade pode ser avaliada usando os orbitais moleculares de fronteira , que podem ser obtidos computacionalmente. Em geral, o átomo que carrega o maior coeficiente orbital no HOMO atua como o nucleófilo, enquanto que no LUMO atua como o eletrófilo. O átomo mais nucleofílico é geralmente, mas nem sempre, o átomo mais rico em elétrons. Em cicloadições 1,3-dipolares, a identidade do par dipolar-dipolarófilo determina se o caráter HOMO ou LUMO do 1,3-dipolo vai dominar (ver discussão sobre orbitais moleculares de fronteira abaixo).
Dipolarófilo
Os dipolarófilos mais comumente usados são alcenos e alcinos. Dipolarófilos contendo heteroátomo , como carbonilas e iminas, também podem sofrer cicloadição 1,3-dipolar. Outros exemplos de dipolarófilos incluem fulerenos e nanotubos , que podem sofrer cicloadição 1,3-dipolar com ileto de azometina na reação de Prato .
Efeitos do solvente
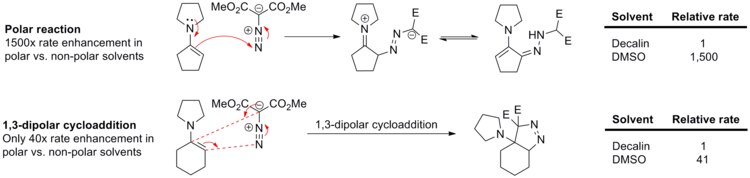
As cicloadições 1,3-dipolares experimentam muito pouco efeito de solvente porque ambos os reagentes e os estados de transição são geralmente apolares. Por exemplo, a taxa de reação entre fenil diazometano e acrilato de etila ou norborneno (ver esquema abaixo) muda apenas ligeiramente com a variação de solventes de ciclohexano para metanol.

A falta de efeitos do solvente na cicloadição 1,3-dipolar é claramente demonstrada na reação entre enaminas e diazomalonato de dimetila (ver esquema abaixo). A reação polar, adição nucleofílica de N-ciclo pen tenil pirrolidina ao composto diazo, ocorre 1.500 vezes mais rápido em DMSO polar do que em decalina apolar . Por outro lado, um análogo próximo desta reação, N-ciclo hex enil pirrolidina 1,3-dipolar cicloadição a dimetil diazomalonato, é acelerado apenas 41 vezes em DMSO em relação à decalina.
Teoria de orbitais moleculares de fronteira
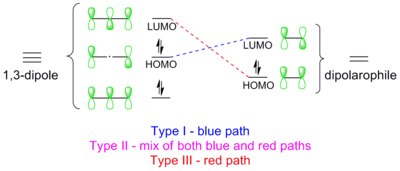
As cicloadições 1,3-dipolares são reações pericíclicas, que obedecem às regras de Dewar-Zimmerman e às regras de Woodward-Hoffmann . No tratamento Dewar-Zimmerman, a reação prossegue através de um estado de transição de Huckel de 6 elétrons de 5 centros, nós zero para este diagrama orbital molecular em particular. No entanto, cada orbital pode receber um sinal aleatoriamente para chegar ao mesmo resultado. No tratamento de Woodward-Hoffmann, orbitais moleculares de fronteira (FMO) do 1,3-dipolo e do dipolarófilo se sobrepõem na maneira permitida pela simetria π 4 s + π 2 s . Essa sobreposição orbital pode ser alcançada de três maneiras: tipo I, II e III. A via dominante é aquela que possui a menor lacuna de energia HOMO-LUMO.
Tipo I
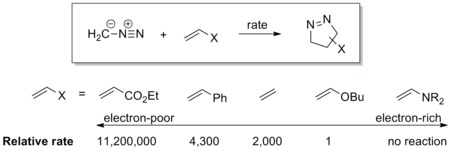
O dipolo tem um HOMO alto que se sobrepõe ao LUMO do dipolarófilo. Um dipolo desta classe é referido como um dipolo controlado por HOMO ou um dipolo nucleofílico , que inclui ileto de azometina , ileto de carbonila , ileto de nitrila , azometina imina , carbonil imina e diazoalcano . Esses dipolos adicionam-se prontamente aos alcenos eletrofílicos. Os grupos retiradores de elétrons (EWG) no dipolarófilo acelerariam a reação diminuindo o LUMO, enquanto os grupos doadores de elétrons (EDG) desacelerariam a reação aumentando o HOMO. Por exemplo, a escala de reatividade do diazometano contra uma série de dipolarófilos é mostrada no esquema abaixo. O diazometano reage com o acrilato de etila pobre em elétrons mais de um milhão de vezes mais rápido do que o éter butil vinílico rico em elétrons.
Esse tipo se assemelha à reação de Diels-Alder de demanda normal de elétrons, na qual o dieno HOMO se combina com o dienófilo LUMO.
Tipo II
O HOMO do dipolo pode emparelhar com o LUMO do dipolarófilo; alternativamente, o HOMO do dipolarófilo pode emparelhar com o LUMO do dipolo. Essa interação bidirecional surge porque a lacuna de energia em qualquer direção é semelhante. Um dipolo desta classe é referido como um dipolo controlado por HOMO-LUMO ou um dipolo ambifílico , que inclui imida de nitrila , nitrona , óxido de carbonila , óxido de nitrila e azida . Qualquer substituinte no dipolarófilo aceleraria a reação, diminuindo a lacuna de energia entre os dois orbitais em interação; ou seja, um EWG diminuiria o LUMO enquanto um EDG aumentaria o HOMO. Por exemplo, as azidas reagem com vários dipolarófilos ricos e pobres em elétrons com reatividades semelhantes (ver escala de reatividade abaixo).

Tipo III
O dipolo tem um LUMO baixo que se sobrepõe ao HOMO do dipolarófilo (indicado por linhas vermelhas tracejadas no diagrama). Um dipolo desta classe é referido como um dipolo controlado por LUMO ou um dipolo eletrofílico , que inclui óxido nitroso e ozônio . EWGs no dipolarófilo desaceleram a reação, enquanto EDGs aceleram a reação. Por exemplo, o ozônio reage com o 2-metilpropeno rico em elétrons cerca de 100.000 vezes mais rápido do que o tetracloroeteno pobre em elétrons (veja a escala de reatividade abaixo).
Este tipo se assemelha à reação de Diels-Alder de demanda inversa de elétrons , na qual o dieno LUMO se combina com o dienófilo HOMO.

Reatividade
Processos combinados como a cicloadição 1,3 requerem um estado de transição altamente ordenado (alta entropia negativa de ativação) e apenas requisitos de entalpia moderados. Usando experimentos de reação de competição, taxas relativas de adição para diferentes reações de cicloadição oferecem resultados gerais sobre fatores de reatividade.
- A conjugação , especialmente com grupos aromáticos, aumenta a taxa de reação por estabilização do estado de transição. Durante a transição, as duas ligações sigma estão sendo formadas em taxas diferentes, o que pode gerar cargas parciais no estado de transição que podem ser estabilizadas por distribuição de carga em substituintes conjugados.
- Dipolarófilos mais polarizáveis são mais reativos porque nuvens difusas de elétrons são mais adequadas para iniciar o fluxo de elétrons.
- Dipolarófilos com alta deformação angular são mais reativos devido ao aumento da energia do estado fundamental.
- O aumento do impedimento estérico no estado de transição, como resultado de reagentes não impedidos, reduz drasticamente a taxa de reação.
- Os heterodipolarófilos adicionam mais lentamente, se o fizerem, em comparação com os C, C-diapolarófilos devido a um menor ganho na energia da ligação sigma para compensar a perda de uma ligação pi durante o estado de transição.
- O isomerismo do dipolarófilo afeta a taxa de reação devido aos estéricos. os isômeros trans são mais reativos (o trans- estilbeno adicionará difenil (nitrila imida) 27 vezes mais rápido do que o cis- estilbeno) porque durante a reação, o ângulo de ligação de 120 ° encolhe para 109 °, trazendo os substituintes cis eclipsantes entre si para aumentar choque estérico.

Estereoespecificidade
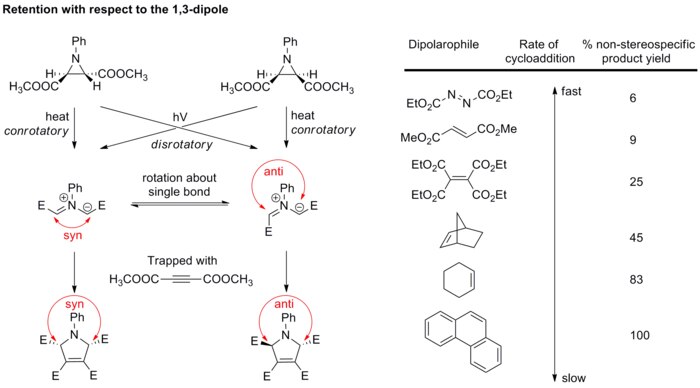
As cicloadições 1,3-dipolares geralmente resultam na retenção da configuração em relação ao 1,3-dipolo e ao dipolarófilo. Esse alto grau de estereoespecificidade é um forte suporte para os mecanismos de reação combinados sobre os graduais. Como mencionado anteriormente, muitos exemplos mostram que as reações foram graduais, apresentando, portanto, estereoespecificidade parcial ou nenhuma estereoespecificidade.
Com relação ao dipolarófilo
Os substituintes cis no alceno dipolarofílico terminam em cis , e os substituintes trans terminam em trans no composto cíclico de cinco membros resultante (ver esquema abaixo).

Com relação ao dipolo
Geralmente, a estereoquímica do dipolo não é uma grande preocupação porque apenas alguns dipolos poderiam formar centros estereogênicos , e as estruturas de ressonância permitem a rotação da ligação que embaralha a estereoquímica. No entanto, o estudo dos iletos de azometina verificou que a cicloadição também é estereoespecífica em relação ao componente dipolo. Os iletos de azometina diastereopuros são gerados pela abertura do anel eletrocíclico das aziridinas e, em seguida, rapidamente capturados com dipolarófilos fortes antes que a rotação da ligação possa ocorrer (ver esquema abaixo). Se dipolarófilos mais fracos forem usados, as ligações no dipolo têm a chance de girar, resultando em estereoespecificidade de cicloadição prejudicada.
Esses resultados em conjunto confirmam que a cicloadição 1,3-dipolar é estereoespecífica, dando retenção tanto do 1,3-dipolo quanto do dipolarófilo.
Diastereosseletividade
Quando dois ou mais estereocentros são gerados durante a reação, estados de transição diastereoméricos e produtos podem ser obtidos. Na cicloadição de Diels-Alder, a diastereosseletividade endo devido a interações orbitais secundárias é geralmente observada. Em cicloadições 1,3-dipolares, entretanto, duas forças influenciam a diastereosseletividade: a interação π atrativa (lembrando as interações orbitais secundárias na cicloadição de Diels-Alder) e a interação estérica repulsiva . Infelizmente, essas duas forças muitas vezes se cancelam, causando diastereosseleção pobre na cicloadição 1,3-dipolar.
Exemplos de cicloadições 1,3-dipolares diastereosseletivas controladas por substrato são mostrados abaixo. A primeira é a reação entre o N-benzileto de benzonitrila e o acrilato de metila . No estado de transição, os grupos fenil e metil éster empilham-se para dar a substituição cis como o produto final exclusivo da pirrolina . Esta interação π favorável compensa a repulsão estérica entre os grupos fenil e metil éster. O segundo é a reação entre nitrona e diidrofurano . A exo- seletividade é obtida para minimizar a repulsão estérica. A última é a reação intramolecular de ileto de azometina com alceno. A diastereosseletividade é controlada pela formação de um sistema de anel cis - fundido menos tenso .

Cicloadição 1,3-dipolar direcionada
A trajetória da cicloadição pode ser controlada para atingir uma reação diastereosseletiva. Por exemplo, os metais podem quelar para o dipolarófilo e o dipolo de entrada e direcionar a cicloadição seletivamente em uma face. O exemplo abaixo mostra a adição de óxido de nitrila a um álcool alílico enantiomericamente puro na presença de um íon magnésio. A conformação mais estável do alceno coloca o grupo hidroxila acima do plano do alceno. O magnésio então se quela para o grupo hidroxila e o átomo de oxigênio do óxido de nitrila. A cicloadição, portanto, vem da face superior seletivamente.
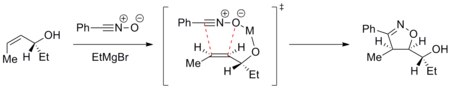
Essa diastereodirecção foi aplicada na síntese de epotilonas .

Regiosseletividade
Para pares dipolarofílicos assimétricos, dois produtos regioisoméricos são possíveis. Ambas as electrónicas / estereoeletrônicas factores estéricos e contribuir para a regiosselectividade da cicloadição 1,3-dipolar.
Efeito eletrônico / estereeletrônico
A interação eletrônica dominante é a combinação entre o maior HOMO e o maior LUMO. Portanto, a regiosseletividade é governada pelos átomos que carregam os maiores coeficientes orbitais HOMO e LUMO.
Por exemplo, considere a cicloadição de diazometano a três dipolarófilos: acrilato de metila , estireno ou cinamato de metila . O carbono do diazometano carrega o maior HOMO, enquanto os carbonos olefínicos finais do acrilato de metila e do estireno carregam o maior LUMO. Portanto, a cicloadição dá a substituição na posição C-3 regiosseletivamente. Para o cinamato de metila, os dois substituintes (Ph vs COOMe) competem na retirada de elétrons do alceno. O carboxila é o melhor grupo de retirada de elétrons, fazendo com que o carbono β seja mais eletrofílico. Assim, a cicloadição produz o grupo carboxila em C-3 e o grupo fenila em C-4 regiosseletivamente.

Efeito estérico
Os efeitos estéricos podem cooperar ou competir com os efeitos eletrônicos mencionados anteriormente. Às vezes, os efeitos estéricos superam completamente a preferência eletrônica, dando exclusivamente o regioisômero oposto.
Por exemplo, diazometano geralmente adiciona acrilato de metila para dar 3-carboxil pirazolina . No entanto, ao colocar mais demandas estéricas no sistema, começamos a observar as 4-carboxil pirazolinas isoméricas. A proporção desses dois regioisômeros depende das demandas estéricas. No extremo, aumentar o tamanho de hidrogênio para t-butil muda a regiosseletividade de 100% de 3-carboxil para 100% de substituição de 4-carboxil.

Aplicações sintéticas
As cicloadições 1,3-dipolares são caminhos importantes para a síntese de muitos heterociclos de 5 membros importantes, como triazóis , furanos , isoxazóis , pirrolidinas e outros. Além disso, alguns cicloaddutos podem ser clivados para revelar o esqueleto linear, fornecendo outra rota para a síntese de compostos alifáticos . Estas reações são tremendamente úteis também porque são estereoespecíficas, diastereosseletivas e regiosseletivas. Vários exemplos são fornecidos abaixo.
Óxidos de nitrilo
A cicloadição 1,3-dipolar com óxidos de nitrila é uma reação aldólica mascarada amplamente utilizada . A cicloadição entre um óxido de nitrila e um alceno produz o produto de isoxazolina cíclica, enquanto a reação com um alcino produz o isoxazol. Tanto as isoxazolinas como os isoxazoles podem ser clivados por hidrogenação para revelar produtos β-hidroxicarbonil do tipo aldol ou β-dicarbonil do tipo Claisen , respectivamente.
A cicloadição de óxido de nitrila-alquino seguida de hidrogenação foi utilizada na síntese de Miyakolide como ilustrado na figura abaixo.

Iletos de carbonila
As reações de cicloadição 1,3-dipolar surgiram como ferramentas poderosas na síntese de estruturas e moléculas cíclicas complexas para estudos medicinais, biológicos e mecanísticos. Entre eles, reações de cicloadição [3 + 2] envolvendo carbonil iletos têm sido amplamente empregadas para gerar moléculas cíclicas de cinco membros contendo oxigênio.
Preparação de carbonil iletos para reações de cicloadição 1,3-dipolar
Os iletos são considerados heteroátomos carregados positivamente conectados a átomos de carbono carregados negativamente, que incluem iletos de sulfônio , tiocarbonila , oxônio , nitrogênio e carbonila . Existem vários métodos para gerar carbonil iletos, que são intermediários necessários para gerar estruturas em anel de cinco membros contendo oxigênio, para reações de cicloadição [3 + 2].
Síntese de carbonil iletos a partir de derivados de diazometano por fotocatálise
Um dos primeiros exemplos de síntese de ileto de carbonil envolve a fotocatálise . A fotólise de diazotetraquis (trifluorometil) ciclopentadieno * (DTTC) na presença de tetrametilureia pode gerar o carbonil ileto por um ataque nucleofílico intermolecular e subsequente aromatização da fração DTTC. Este foi isolado e caracterizado por cristalografia de raios-X devido à estabilidade conferida pela aromaticidade, grupos trifluorometil que retiram elétrons e os grupos dimetilamina doadores de elétrons . Carbonil ileto estáveis dipolos podem então ser utilizados em reacções de cicloadição [3 + 2] com dipolarófilos .

Outro exemplo inicial de síntese de carbonil ileto por fotocatálise foi relatado por Olah et al . O dideuteriodiazometano foi fotolisado na presença de formaldeído para gerar o carbonil ileto de dideuterioformaldeído.

Síntese de carbonil iletos a partir de hidroxipironas por transferência de prótons
Os iletos de carbonila podem ser sintetizados por catálise ácida de hidroxi-3-pironas na ausência de um catalisador de metal . Ocorre uma tautomerização inicial , seguida pela eliminação do grupo de saída para aromatizar o anel de pirona e gerar o ileto de carbonila. Uma reação de cicloadição com um dipolarófilo forma por último o oxaciclo. Esta abordagem é menos amplamente utilizada devido à sua utilidade limitada e à necessidade de esqueletos de pirona.

5-hidroxi-4-pironas também podem ser usadas para sintetizar carbonil iletos por uma transferência de hidrogênio intramolecular . Após a transferência de hidrogênio, o carbonil ileto pode então reagir com dipolarófilos para formar anéis contendo oxigênio.

Síntese de α-halocarbonil iletos a partir de dihalocarbenos
Os dihalocarbenos também têm sido empregados para gerar iletos de carbonil, explorando a natureza de retirada de elétrons dos dihalocarbenos. Tanto o fenil (triclorometil) mercúrio quanto o fenil (tribromometil) mercúrio são fontes diclorocarbenos e dibromocarbenos , respectivamente. O carbonil ileto pode ser gerado na reação dos dihalocarbenos com cetonas ou aldeídos . No entanto, a síntese de a-halocarbonil iletos também pode levar indesejavelmente à perda de monóxido de carbono e à geração do produto de desoxigenação.

Síntese de carbonil iletos a partir de derivados de diazometano por catálise de metal
Uma abordagem universal para a geração de iletos de carbonila envolve a catálise metálica de compostos α-diazocarbonila, geralmente na presença de catalisadores dicobre ou diródio. Após a liberação do gás nitrogênio e a conversão ao metalocarbeno , uma reação intermolecular com um grupo carbonila pode gerar o ileto de carbonila. A reação de cicloadição subsequente com um alceno ou alcino dipolarófilo pode proporcionar anéis de cinco membros contendo oxigênio. Os catalisadores populares que fornecem rendimentos modestos para a síntese de oxaciclos incluem Rh 2 (OAc) 4 e Cu (acac) 2 .

Mecanismo da reação de cicloadição 1,3-dipolar mediada pela catálise metálica de compostos diazocarbonil
A universalidade e o uso extensivo de reações de cicloadição 1,3-dipolar mediadas por catálise de metal de moléculas de diazocarbonil, para sintetizar anéis de cinco membros contendo oxigênio, tem estimulado um interesse significativo em seu mecanismo. Vários grupos investigaram o mecanismo para expandir o escopo das moléculas sintéticas no que diz respeito à regio e estereo-seletividade . No entanto, devido às altas frequências de rotação dessas reações, os intermediários e o mecanismo permanecem indefinidos. O mecanismo geralmente aceito, desenvolvido pela caracterização de complexos estáveis de rutênio-carbenóide e metalocarbenos de ródio, envolve uma formação inicial de um complexo metal-carbenóide a partir do composto diazo . A eliminação do gás nitrogênio fornece um metalocarbeno. Um ataque nucleofílico intramolecular pelo oxigênio da carbonila regenera o catalisador de metal e forma o ileto de carbonila. O carbonil ileto pode então reagir com um alceno ou alcino, tal como dimetil acetilenodicarboxilato (DMAD) para gerar o oxaciclo.

No entanto, é incerto se o intermediário de metalocarbeno gera o carbonil ileto. Em alguns casos, os metalocarbenos também podem reagir diretamente com os dipolarófilos. Nestes casos, o metalocarbeno, como o diródio (II) tetracarboxilato carbeno, é estabilizado por meio de interações do tipo enolato metálico hiperconjugativo . A reação de cicloadição 1,3-dipolar subsequente ocorre através de um carbonil ileto de complexo metálico transitório. Portanto, um metalocarbeno persistente pode influenciar a estereosseletividade e a regiosseletividade da reação de cicloadição 1,3-dipolar com base na estereoquímica e no tamanho dos ligantes metálicos .
tetracarboxylate_metallocarbene_stabilized_by_%CF%80C-Rh%E2%86%92%CF%80C%3DO_hyperconjugation..png)
O mecanismo da reação de cicloadição 1,3-dipolar entre o dipolo de carbonil ileto e alcinil ou alquenil dipolarófilos foi extensivamente investigado no que diz respeito à regiosseletividade e estereosseletividade. Como os dipolarófilos simétricos têm uma orientação para cicloadição, apenas um regioisômero , mas vários estereoisômeros podem ser obtidos. Pelo contrário, dipolarófilos assimétricos podem ter múltiplos regioisômeros e estereoisômeros. Esses regioisômeros e estereoisômeros podem ser previstos com base na teoria de orbitais moleculares de fronteira (FMO) , interações estéricas e interações estereeletrônicas .

Regiosseletividade da reação de cicloadição 1,3-dipolar mediada pela catálise de metais de compostos diazocarbonil
A regiosseletividade de reações de cicloadição 1,3-dipolar entre dipolos de carbonil ileto e alcinil ou alquenil dipolarófilos é essencial para gerar moléculas com regioquímica definida. A teoria FMO e a análise das lacunas de energia do HOMO-LUMO entre o dipolo e o dipolarófilo podem racionalizar e prever a regiosseletividade dos resultados experimentais. Os homos e LUMOS pode pertencer a um dipolo ou dipolarófilo, para que HOMO dipolo -LUMO dipolarófilo ou HOMO dipolarófilo -LUMO dipolo pode existir interacções. A sobreposição dos orbitais com os maiores coeficientes pode, em última análise, racionalizar e prever os resultados.

A regiosseletividade arquetípica da reação de cicloadição 1,3-dipolar mediada por dipolos de carbonil ileto foi examinada por Padwa e colaboradores. Usando um Rh 2 (OAc) 4 catalisador em benzeno, diazodione foram submetidos a uma reacção de cicloadição 1,3-dipolar com metil propiolato e metilo propargilo éter . A reação com propiolato de metila fornece dois regioisômeros com o principal resultante da interação dipolarófila HOMO dipolo- LUMO , que tem os maiores coeficientes no carbono proximal ao grupo carbonila do ileto de carbonila e no carbono alquino terminal de propiolato de metila. A reacção com metil propargilo éter proporciona um regioisómero resultante da HOMO dipolarófilo -LUMO dipolo interacção, que tem maiores coeficientes no distai de carbono ao grupo carbonilo do ileto de carbonilo e na metil éter propargilo de carbono alcino terminal.

As regiosseletividades das reações de cicloadição 1,3-dipolar mediadas pela catálise metálica de compostos diazocarbonil também podem ser influenciadas pelo metal através da formação de metalocarbenos estáveis. A estabilização do metalocarbeno, por meio de interações do tipo enolato de metal, evitará a formação de iletos de carbonila, resultando em uma reação direta entre o dipolo de metalocarbeno e um alcinil ou alcenil dipolarófilo (ver imagem do metalocarbeno de tetracarboxilato de diródio (II) estabilizado por π C -Rh → π C = O hiperconjugação.). Nesta situação, os ligantes metálicos irão influenciar a regiosseletividade e estereosseletividade da reação de cicloadição 1,3-dipolar.
Estereosseletividade e indução assimétrica da reação de cicloadição 1,3-dipolar mediada por catálise de metais de compostos diazocarbonil
A estereosseletividade das reações de cicloadição 1,3-dipolar entre dipolos de carbonil ileto e alquenil dipolarófilos também foi examinada de perto. Para alcinil dipolarófilos, a estereosseletividade não é um problema, pois carbonos sp 2 relativamente planares são formados, enquanto a regiosseletividade deve ser considerada (veja a imagem dos produtos da reação de cicloadição 1,3-Dipolar entre os dipolos de carbonil ileto e alquenil ou alquinil dipolarófilos). No entanto, para alquenil dipolarófilos, tanto a regiosseletividade quanto a estereosseletividade devem ser consideradas, uma vez que os carbonos sp 3 são gerados nas espécies do produto.
As reações de cicloadição 1,3-dipolar entre dipolos de carbonil ileto e alquenil dipolarófilos podem gerar produtos diastereoméricos . O produto exo é caracterizado com substituintes dipolarófilos sendo cis para a ponte éter do oxaciclo. O produto endo é caracterizado com os substituintes dipolarófilos sendo trans para a ponte éter do oxaciclo. Ambos os produtos podem ser gerados através pericíclicas transições estados envolvendo concertadas processos assíncronos síncronos ou concertadas.
Um dos primeiros exemplos conferiu estereosseletividade em termos de produtos endo e exo com catalisadores de metal e ácidos de Lewis. As reações apenas com o catalisador metálico Rh 2 (OAc) 4 preferem o produto exo, enquanto as reações com o ácido de Lewis adicional Yb (OTf) 3 preferem o produto endo . A seletividade endo observada para reações de cicloadição ácida de Lewis é atribuída à sobreposição orbital otimizada dos sistemas π da carbonila entre o dipolarófilo coordenado por Yb (Otf) 3 (LUMO) e o dipolo (HOMO). Após muitas investigações, duas abordagens principais para influenciar a estereosseletividade de cicloadições de ileto de carbonil foram desenvolvidas para explorar a quiralidade de catalisadores metálicos e ácidos de Lewis.


A primeira abordagem emprega catalisadores de metal quirais para modular a estereosseletividade endo e exo . Os catalisadores quirais, em particular Rh 2 [( S ) -DOSP] 4 e Rh 2 [( S ) -BPTV] 4 podem induzir indução assimétrica modesta e foram usados para sintetizar o agente antifúngico ácido pseudolárico A. Este é um resultado da catalisador de metal quiral que permanece associado ao ileto de carbonila durante a cicloadição, o que confere seletividade facial. No entanto, os mecanismos exatos ainda não são totalmente compreendidos.

A segunda abordagem emprega um catalisador ácido de Lewis quiral para induzir a estereosseletividade facial após a geração do ileto de carbonila usando um catalisador metálico aquiral. Acredita-se que o catalisador ácido de Lewis quiral coordene com o dipolarófilo, o que diminui o LUMO do dipolarófilo, ao mesmo tempo que leva à enantiosseletividade .

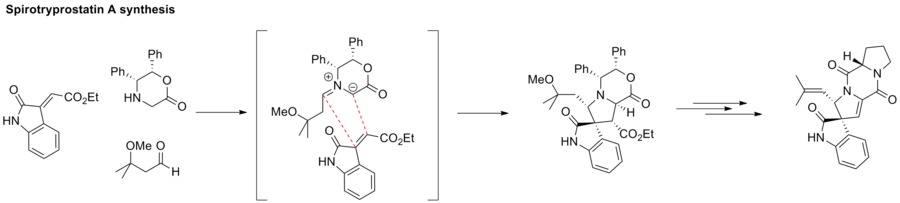
Iletos de azometina
A cicloadição 1,3-dipolar entre um ileto de azometina e um alceno fornece uma estrutura azacíclica, como a pirrolidina . Esta estratégia tem sido aplicada à síntese de espirotriprostatina A.
Ozônio
A ozonólise é uma reação orgânica muito importante. Alcenos e alcinos podem ser clivados por ozonólise para dar produtos de aldeído , cetona ou ácido carboxílico .
Aplicações biológicas
A cicloadição 1,3-dipolar entre azidas orgânicas e alcinos terminais (ou seja, a cicloadição de Huisgen ) tem sido amplamente utilizada para bioconjugação .
Catálise de cobre
A reação de Huisgen geralmente não ocorre prontamente em condições moderadas. Meldal et al. e Sharpless et al. desenvolveu de forma independente uma versão catalisada por cobre (I) da reação de Huisgen, CuAAC (para cicloadição azida-alcina catalisada por cobre), que prossegue muito prontamente em condições suaves, incluindo fisiológicas ( pH neutro , temperatura ambiente e solução de água ). Esta reação também é bioortogonal : azidas e alcinos estão geralmente ausentes dos sistemas biológicos e, portanto, essas funcionalidades podem ser reagidas quimiosseletivamente, mesmo no contexto celular . Eles também não reagem com outros grupos funcionais encontrados na natureza, portanto, não perturbam os sistemas biológicos. A reação é tão versátil que é denominada química do "clique" . Embora o cobre (I) seja tóxico , muitos ligantes protetores foram desenvolvidos para reduzir a citotoxicidade e melhorar a taxa de CuAAC, permitindo que seja usado em estudos in vivo .

Por exemplo, Bertozzi et al. relataram a incorporação metabólica de sacarídeos funcionalizados com azida no glicano da membrana celular e subsequente marcação com conjugado fluoróforo- alquino. A membrana celular agora marcada com fluorescência pode ser visualizada no microscópio .

Cicloadição promovida por tensão
Para evitar a toxicidade do cobre (I), Bertozzi et al. desenvolveram a cicloadição de azida-alcino promovida por cepa (SPAAC) entre a azida orgânica e o ciclo- octino tenso . A distorção do ângulo do ciclo-octino ajuda a acelerar a reação, reduzindo a tensão de ativação e aumentando as interações, permitindo assim que seja usado em condições fisiológicas sem a necessidade do catalisador.

Por exemplo, Ting et al. introduziu uma funcionalidade azido em proteínas específicas na superfície celular usando uma enzima ligase . A proteína marcada com azida é então marcada com o conjugado ciclo-octino-fluoróforo para produzir uma proteína marcada com fluorescência.

Referências
- ^ Huisgen, Rolf (1963). "1.3-Dipolare Cycloadditionen Ruckschau und Ausblick". Angewandte Chemie . 75 (13): 604–637. doi : 10.1002 / ange.19630751304 .
- ^ a b Huisgen, Rolf (novembro de 1963). "Kinetics and Mechanism of 1,3-Dipolar Cycloadditions". Angewandte Chemie International Edition . 2 (11): 633–645. doi : 10.1002 / anie.196306331 .
- ^ Firestone, R (1968). "Mecanismo de cicloadições 1,3-dipolares". Journal of Organic Chemistry . 33 (6): 2285–2290. doi : 10.1021 / jo01270a023 .
- ^ Huisgen, Rolf (1976). "Cicloadições 1,3-Dipolares. 76. Natureza combinada de cicloadições 1,3-dipolares e a questão dos intermediários diradicais". Journal of Organic Chemistry . 41 (3): 403–419. doi : 10.1021 / jo00865a001 .
- ^ Mloston, G .; Langhals, E .; Huisgen, Rolf (1986). "First Two-Step 1,2-Dipolar Cycloadditons: Nonstereospecificity". Geléia. Chem. Soc. 108 (20): 6401–66402. doi : 10.1021 / ja00280a053 .
- ^ Seyyed Amir, Siadati (2015). "Um exemplo de um mecanismo passo a passo para a cicloadição 1,3-dipolar livre de catalisador entre um óxido de nitrila e um alceno rico em elétrons". Letras de tetraedro . 56 (34): 4857–4863. doi : 10.1016 / j.tetlet.2015.06.048 .
- ^ Huisgen, Rolf (1963). "1,3-Dipolar Cycloadditions. Past and Future". Angewandte Chemie International Edition . 2 (10): 565–598. doi : 10.1002 / anie.196305651 .
- ^ Cox, A; Thomas, L; Sheridan, J (1958). "Microwave Spectra of Diazomethane and its Deutero Derivatives". Nature . 181 (4614): 1000–1001. Bibcode : 1958Natur.181.1000C . doi : 10.1038 / 1811000a0 . S2CID 4245746 .
- ^ Hilberty, P; Leforestier, C (1978). "Expansão das funções de onda orbital molecular em funções de onda de ligação de valência. Um procedimento simplificado". Journal of the American Chemical Society . 100 (7): 2012–2017. doi : 10.1021 / ja00475a007 .
- ^ McGarrity, JF; Patai, Saul (1978). Basicidade, acidez e ligações de hidrogênio . Grupos Diazônio e Diazo . 1 . pp. 179–230. doi : 10.1002 / 9780470771549.ch6 . ISBN 9780470771549.
- ^ Berner, Daniel; McGarrity, John (1979). "Observação direta do íon metildiazônio em ácido fluorossulfúrico". Journal of the American Chemical Society . 101 (11): 3135–3136. doi : 10.1021 / ja00505a059 .
- ^ Muller, Eugen; Rundel, Wolfgans (1956). "Untersuchungen an Diazomethanen, VI. Mitteil .: Umsetzung von Diazoäthan mit Methyllithium". Chemische Berichte . 89 (4): 1065–1071. doi : 10.1002 / cber.19560890436 .
- ^ Geittner, Jochen; Huisgen, Rolph; Reissig, Hans-Ulrich (1978). "Dependência de solvente das taxas de cicloadição de fenildiazometano e parâmetros de ativação". Heterociclos . 11 : 109-120. doi : 10.3987 / S (N) -1978-01-0109 .
- ^ Huisgen, Rolph; Reissig, Hans-Ulrich; Huber, Helmut; Voss, Sabine (1979). "Compostos α-Diazocarbonil e enaminas - uma dicotomia de vias de reação". Letras de tetraedro . 20 (32): 2987–2990. doi : 10.1016 / S0040-4039 (00) 70991-9 .
- ^ Sustmann, R (1974). Controle de energia orbital da reatividade de cicloadição " . Química pura e aplicada . 40 (4): 569–593. doi : 10.1351 / pac197440040569 .
- ^ Geittner, Jochen; Huisgen, Rolf (1977). "Cinética das reações de cicloadição 1,3-dipolar de diazometano; Uma correlação com energias homo-lumo". Letras de tetraedro . 18 (10): 881–884. doi : 10.1016 / S0040-4039 (01) 92781-9 .
- ^ Huisgen, Rolf; Szeimies, Gunter; Mobius, Leander (1967). "K1.3-Dipolare Cycloadditionen, XXXII. Kinetik der Additionen organischer Azide an CC-Mehrfachbindungen". Chemische Berichte . 100 (8): 2494–2507. doi : 10.1002 / cber.19671000806 .
- ^ Williamson, DG; Cvetanovic, RJ (1968). "Taxas de reações de ozônio-olefina em soluções de tetracloreto de carbono". Journal of the American Chemical Society . 90 (14): 3668–3672. doi : 10.1021 / ja01016a011 .
- ^ Bihlmaier, Werner; Geittner, Jochen; Huisgen, Rolf; ReissigP, Hans-Ulrich (1978). "The Stereospecificity of Diazomethane Cycloadditions" . Heterociclos . 10 : 147–152. doi : 10.3987 / S-1978-01-0147 .
- ^ Huisgen, Rolf; Scheer, Wolfgang; Huber, Helmut (1967). "Stereospecific Conversion of cis-trans Isomeric Aziridines to Open-Chain Azomethine Ylides". Journal of the American Chemical Society . 89 (7): 1753–1755. doi : 10.1021 / ja00983a052 .
- ^ Dahmen, Alexander; Hamberger, Helmut; Huisgen, Rolf; Markowski, Volker (1971). "Abertura do anel conrotatório de óxidos de cianostilbeno para iletos de carbonila". Journal of the Chemical Society D: Chemical Communications (19): 1192–1194. doi : 10.1039 / C29710001192 .
- ^ Padwa, Albert; Smolanoff, Joel (1971). "Photocycloaddition of arylazirenes with electron-deficient olefins". Journal of the American Chemical Society . 93 (2): 548–550. doi : 10.1021 / ja00731a056 .
- ^ Iwashita, Takashi; Kusumi, Takenori; Kakisawa, Hiroshi (1979). "A Synthesis of dl-isoretronecanol" . Cartas de química . 8 (11): 1337–1340. doi : 10.1246 / cl.1979.1337 .
- ^ Wang, Chia-Lin; Ripka, William; Confalone, Pat (1984). "Uma síntese curta e estereoespecífica de (±) -α-lycorano". Letras de Tetraedro . 25 (41): 4613–4616. doi : 10.1016 / S0040-4039 (01) 91213-4 .
- ^ Kanemasa, Shuji (2002). "Metal-Assisted Stereocontrol of 1,3-Dipolar Cycloaddition Reactions". Synlett . 2002 (9): 1371–1387. doi : 10.1055 / s-2002-33506 .
- ^ Bode, Jeffrey; Carreira, Erick (2011). "Stereoselective Syntheses of Epothilones A and B via Directed Nitrile Oxide Cycloaddition". Journal of the American Chemical Society . 123 (15): 3611–3612. doi : 10.1021 / ja0155635 . PMID 11472140 .
- ^ Vsevolod V. Rostovtsev; Luke G. Green; Valery V. Fokin; K. Barry Sharpless (2002). "A Stepwise Huisgen Cycloaddition Process: Copper (I) -Catalyzed Regioselective Ligation of Azides and Terminal Alkynes". Angewandte Chemie International Edition . 41 (14): 2596–22599. doi : 10.1002 / 1521-3773 (20020715) 41:14 <2596 :: AID-ANIE2596> 3.0.CO; 2-4 . PMID 12203546 .
- ^ Caramella, Pierluigi; Houk, KN (1976). "Geometries of nitrilium betaaines. O esclarecimento de reações aparentemente anômalas de 1,3-dipolos". Journal of the American Chemical Society . 98 (20): 6397–6399. doi : 10.1021 / ja00436a062 .
- ^ Caramella, Pierluigi; Gandour, Ruth W .; Hall, Janet A .; Deville, Cynthia G .; Houk, KN (1977). "Uma derivação das formas e energias dos orbitais moleculares de 1,3-dipolos. Otimizações geométricas dessas espécies por MINDO / 2 e MINDO / 3". Journal of the American Chemical Society . 99 (2): 385–392. doi : 10.1021 / ja00444a013 .
- ^ Huisgen, Rolf (novembro de 1963). "Kinetics and Mechanism of 1,3-Dipolar Cycloadditions". Angewandte Chemie International Edition . 2 (11): 633–645. doi : 10.1002 / anie.196306331 .
- ^ Padwa, Albert (1983). 1,3-Dipolar Cycloaddition Chemistry . General Heterocyclic Chemistry Series. 1 . Estados Unidos da América: Wiley-Interscience. pp. 141–145. ISBN 978-0-471-08364-1.
- ^ Koszinowski, J. (1980). tese ( tese de doutorado).
- ^ Evans, David; Ripin, David; Halstead, David; Campos, Kevin (1999). "Synthesis and Absolute Stereochemical Assignment of (+) - Miyakolide". Journal of the American Chemical Society . 121 (29): 6816–6826. doi : 10.1021 / ja990789h .
- ^ Reações sintéticas de ligações M = C e M = N: formação de Ylide, rearranjo e cicloadição 1,3-dipolar; Hiyama, TW, J., Ed.; Elsevier, 2007; Vol. 11
- ^ Padwa, Albert .; Hornbuckle, Susan F. (1991). "Formação de Ylide a partir da reação de carbenos e carbenóides com pares solitários de heteroátomo". Revisões químicas . 91 (3): 263–309. doi : 10.1021 / cr00003a001 .
- ^ a b Janulis, Eugene P .; Arduengo, Anthony J. (1983). "Estrutura de um ileto de carbonil estabilizado eletronicamente". Journal of the American Chemical Society . 105 (18): 5929–5930. doi : 10.1021 / ja00356a044 .
- ^ Prakash, GKS; Ellis, RW; Felberg, JD; Olah, GA Formaldehyde 0-Methylide, [CH2 = O + -CH2 : The Parent Carbonyl Ylide] J Am Chem Soc 1986, 108, 1341.
- ^ Sammes, PG; Street, LJ Intra molecular Cyclo adions with Oxido pyrylium Ylides J. Chem. Soc., Chem. Comum. 1982, 1056.
- ^ Garst, ME; McBride, BJ; Douglass III, JG Intramolecular cycloadditions with 2- (ω-alquenil) -5-hidroxi-4-pironas Tetrahedron Lett. 1983, 24, 1675.
- ^ Gisch, John F .; Landgrebe, John A. (1985). "Diclorocarbeno da pirólise flash de vácuo de trimetil (triclorometil) silano. Observação possível de 1,1-dicloro-3-fenilcarbonil ileto" . The Journal of Organic Chemistry . 50 (12): 2050–2054. doi : 10.1021 / jo00212a009 .
- ^ Huan, Zhenwei; Landgrebe, John A .; Peterson, Kimberly (1983). "Dibromocarbonil iletos. Desoxigenação de aldeídos e cetonas por dibromocarbeno". The Journal of Organic Chemistry . 48 (24): 4519–4523. doi : 10.1021 / jo00172a015 .
- ^ Martin, Charles W .; Lund, Paul R .; Rapp, Erich; Landgrebe, John A. (1978). "Carbonil iletos halogenados nas reações de precursores de dihalocarbeno mercurial com benzaldeídos substituídos". The Journal of Organic Chemistry . 43 (6): 1071–1076. doi : 10.1021 / jo00400a009 .
- ^ Hodgson, DM; Bruckl, T .; Glen, R .; Labande, AH; Selden, DA; Dossetter, AG; Redgrave, AJ Catalytic enantioselective intermolecular cycloadditions of 2-diazo-3,6-diketoester-dered carbonyl ylides with alquen dipolarophiles Proceedings of the National Academy of Sciences dos Estados Unidos da América 2004, 101, 5450.
- ^ Padwa, Albert; Hertzog, Donald L .; Nadler, William R. (1994). "Intramolecular Cycloaddition of Isomunchnone Dipoles to Heteroaromatic .pi.-Systems". The Journal of Organic Chemistry . 59 (23): 7072–7084. doi : 10.1021 / jo00102a037 .
- ^ Hamaguchi, M .; Ibata, T. New Type of Mesoionic System. 1,3-Dipolar Cycloaddition of Isomunchnon With Ethylenic Compounds Chem Lett 1975, 499.
- ^ Park, Soon-Bong; Sakata, Naoya; Nishiyama, Hisao (1996). "Aryloxycarbonylcarbene Complexes of Bis (oxazolinyl) pyridineruthenium as Active Intermediates in Asymmetric Catalytic Cyclopropanations". Chemistry - A European Journal . 2 (3): 303–306. doi : 10.1002 / chem.19960020311 .
- ^ Snyder, James P .; Padwa, Albert; Stengel, Thomas; Arduengo, Anthony J .; Jockisch, Alexander; Kim, Hyo-Joong (2001). "A Stable Dirhodium Tetracarboxylate Carbenoid: Crystal Structure, Bonding Analysis, and Catalysis". Journal of the American Chemical Society . 123 (45): 11318–11319. doi : 10.1021 / ja016928o . PMID 11697986 .
- ^ a b Hodgson, DM; Pierard, FYTM; Stupple, PA Catalytic enantioselective rearranjements and cycloadditions envolvendo iletos de compostos diazo Chem Soc Rev 2001, 30, 50.
- ^ Yoshikai, Naohiko; Nakamura, Eiichi (2003). "Theoretical Studies on Diastereo- and Enantioselective Rhodium-Catalyzed Cyclization of Diazo Compoundvia Intramolecular C-H Bond Insertion". Síntese e catálise avançadas . 345 (910): 1159–1171. doi : 10.1002 / adsc.200303092 .
- ^ Nakamura, Eiichi; Yoshikai, Naohiko; Yamanaka, Masahiro (2002). "Mecanismo de ativação da ligação C-H / reação de formação da ligação C-C entre o composto diazo e o alcano catalisado por tetracarboxilato de diródio". Journal of the American Chemical Society . 124 (24): 7181–7192. doi : 10.1021 / ja017823o . PMID 12059244 .
- ^ Costantino, G .; Rovito, R .; Macchiarulo, A .; Pellicciari, R. Estrutura dos intermediários metal-carbenóide derivados da decomposição mediada por tetracarboxilato de diródio (II) de compostos α-diazocarbonil: um estudo DFT J Mol Struc-Theochem 2002, 581, 111.
- ^ a b c d M. Hodgson, D .; H. Labande, A .; Muthusamy, S. In Organic Reactions; John Wiley & Sons, Inc .: 2004.
- ^ Suga, Hiroyuki; Ebiura, Yasutaka; Fukushima, Kazuaki; Kakehi, Akikazu; Baba, Toshihide (2005). "Efficient Catalytic Effects of Lewis Acids in the 1,3-Dipolar Cycloaddition Reactions of Carbonyl Ylides with Imines". The Journal of Organic Chemistry . 70 (26): 10782–10791. doi : 10.1021 / jo051743b . PMID 16356001 .
- ^ a b Padwa, Albert; Fryxell, Glen E .; Zhi, Lin (1990). "Reação em tandem de ciclização-cicloadição de carbenóides de ródio. Âmbito e detalhes mecanísticos do processo". Journal of the American Chemical Society . 112 (8): 3100–3109. doi : 10.1021 / ja00164a034 .
- ^ Houk, KN; Sims, Joyner .; Duke, RE; Strozier, RW; George, John K. (1973). "Orbitais moleculares de fronteira de 1,3 dipolos e dipolarófilos". Journal of the American Chemical Society . 95 (22): 7287–7301. doi : 10.1021 / ja00803a017 .
- ^ Houk, KN; Rondan, Nelson G .; Santiago, Cielo; Gallo, Catherine J .; Gandour, Ruth Wells; Griffin, Gary W. (1980). "Estudos teóricos das estruturas e reações de carbonil iletos substituídos". Journal of the American Chemical Society . 102 (5): 1504–1512. doi : 10.1021 / ja00525a006 .
- ^ Padwa, Albert; Weingarten, M. David (1996). "Processos em Cascata de Carbenoides Metálicos". Revisões químicas . 96 (1): 223–270. doi : 10.1021 / cr950022h . PMID 11848752 .
- ^ Padwa, Albert; Austin, David J. (1996). "Ligand-Induced Selectivity in the Rhodium (II) -Catalyzed Reactions of α-Diazo Carbonyl Compounds †". The Journal of Organic Chemistry . 61 : 63–72. doi : 10.1021 / jo951576n .
- ^ Suga, H .; Kakehi, A .; Ito, S .; Inoue, K .; Ishida, H .; Ibata, T. Stereocontrol in a Ytterbium Triflate-Catalyzed 1,3-Dipolar Cycloaddition Reaction of Carbonyl Ylide com N-Substituted Maleimides and Dimethyl Fumarate B Chem Soc Jpn 2001, 74, 1115.
- ^ Geng, Zhe; Chen, Bin; Chiu, Pauline (2006). "Total Synthesis of Pseudolaric Acid A". Angewandte Chemie International Edition . 45 (37): 6197–6201. doi : 10.1002 / anie.200602056 . PMID 16906616 .
- ^ Suga, Hiroyuki; Inoue, Kei; Inoue, Shuichi; Kakehi, Akikazu; Shiro, Motoo (2005). "Quiral 2,6-Bis (oxazolinyl) pyridine-Rare Earth Metal Complexes as Catalysts for Highly Enantioselective 1,3-Dipolar Cycloaddition Reactions of 2-Benzopyrylium-4-olates". The Journal of Organic Chemistry . 70 (1): 47–56. doi : 10.1021 / jo049007f . PMID 15624905 .
- ^ Onishi, Tomoyuki; Sebahar, Paul; Williams, Robert (2003). "Concise, Asymmetric Total Synthesis of Spirotryprostatin A". Cartas orgânicas . 5 (17): 3135–3137. doi : 10.1021 / ol0351910 . PMID 12917000 .
- ^ Tornoe, cristão; Christensen, Caspar; Meldal, Morten (2002). "Peptidotriazoles on Solid Phase: [1,2,3] -Triazoles by Regiospecific Copper (I) -Catalyzed 1,3-Dipolar Cycloadditions of Terminal Alkynes to Azides". Journal of Organic Chemistry . 67 (9): 3057–3064. doi : 10.1021 / jo011148j . PMID 11975567 .
- ^ Rostovtsev, Vsevolod; Green, Luke; Fokin, Valery; Sharpless, Barry K. (2002). "A Stepwise Huisgen Cycloaddition Process: Copper (I) -Catalyzed Regioselective Ligation of Azides and Terminal Alkynes". Angewandte Chemie International Edition . 41 (14): 2596–2599. doi : 10.1002 / 1521-3773 (20020715) 41:14 <2596 :: AID-ANIE2596> 3.0.CO; 2-4 . PMID 12203546 .
- ^ Besanceney-Webler, Christen; Jiang, Hao; Zheng, Tianqing; Feng, Lei; Soriano del Amo, David; Wang, Wei; Klivansky, Liana M .; Marlow, Florence L .; Liu, Yi; Wu, Peng (2011). "Aumentando a eficácia das reações de clique bioortogonal para bioconjugação: um estudo comparativo" . Angewandte Chemie International Edition . 50 (35): 8051–8056. doi : 10.1002 / anie.201101817 . PMC 3465470 . PMID 21761519 .
- ^ Breidenbach, Mark; Gallagher, Jennifer; King, David; Esperto, Brian; Wu, Peng; Bertozzi, Carolyn (2010). "Marcação metabólica direcionada de N-glicanos de levedura com açúcares não naturais" . Anais da Academia Nacional de Ciências dos Estados Unidos da América . 107 (9): 3988–3993. Bibcode : 2010PNAS..107.3988B . doi : 10.1073 / pnas.0911247107 . PMC 2840165 . PMID 20142501 .
- ^ Agard, Nicholas; Prescher, Jennifer; Bertozzi, Carolyn (2004). "A Strain-Promoted [3 + 2] Azide-Alkyne Cycloadition for Covalent Modification of Biomolecules in Living Systems". Journal of the American Chemical Society . 126 (46): 15046-15047. doi : 10.1021 / ja044996f . PMID 15547999 .
- ^ Fernandez-Suarez, Marta; Baruah, Hemanta; Martinez-Hernandez, Laura; Xie, Kathleen; Baskin, Jeremy; Bertozzi, Carolyn; Ting, Alice (2007). "Redirecionando a ligase do ácido lipóico para a marcação de proteínas da superfície celular com sondas de pequenas moléculas" . Nature Biotechnology . 25 (12): 1483–1487. doi : 10.1038 / nbt1355 . PMC 2654346 . PMID 18059260 .